
Introduction
N-coumaroyldopamine is a novel phenylpropenic acid derivative extracted from Theobroma cacao and was recently demonstrated to agonise the beta-2 receptor en vitro (1, 2). The beta-2 receptor is the most effective mechanism for the induction of fat loss in humans, and the search for natural beta-2 agonists has developed renewed interest from supplement manufacturers.
Pharmacokinetics
N-coumaroyldopamine is highly susceptible to metabolism by Catechol-O-Methyl-Transferase (COMT) due to its 2 meta-phenolic hydroxyl substituents. COMT can also methylate the para positions, but with lower fidelity due to regioselectivity of the COMT isoforms. This enzyme is found in the gastrointestinal mucusoa, the liver, and the periphery, and is a very effective at deactivating catecholamines. Similarly, the phenolic hydroxyls are subject to Phase II metabolism in which the -OH groups are conjugated to more water soluble substituents for more rapid excretion.
Dobutamine, a structurally similar pharmaceutical drug used to increase cardiac contractility, is designed strickly for parental or intravenous administration due to its exceptionally poor bioavailability.
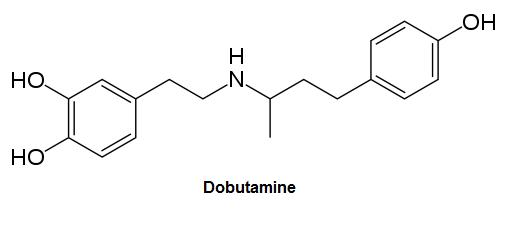
Dopamine Prodrug
As mentioned above, another of N-Coumaroyldopamine's metabolic pathways involves hydrolysis of its amide bond.

Due to the ubiquity of biological hydrolases, the main metabolites of N-Coumaroyldopamine will be Dopamine and a cinnamic acid analogue.

Pharmacodynamics
In the study which examined the adrenergic potential of various Theobroma cacao constituents, N-Coumaroyldopamine was demonstrated to possess highly specific beta-2 adrenergic potential. This is consistent with the literature with regards to the Structure Activity Relationship (SAR) of catecholamine pharmacodynamics. In its unmetabolized form, N-Coumaroyldopamine possesses (1) a meta-OH substituent which allows stronger interaction with the adrenergic receptor and (2) a bulky, fairly nonpolar, substituent coming off the nitrogen. The latter is responsible for increased beta receptor affinity (See Image below).

Summary
N-Coumaroyldopamine is a highly selective beta2 agonist only in its unmetabolized form. Unfortunately, it is exceptionally vulnerable to multiple metabolic processes which makes its ability to actually reach systemic circulation highly unlikely. As a dopamine prodrug, N-coumaroyldopamine will likely also fall short. Successful dopamine prodrugs, like Docarpamine, are generally designed to be resistant to COMT and Phase II metabolism, whereas N-coumaroyldopamine is vulnerable to both.

Moreover, the 4 hydroxyl-phenolic substituents makes the parent compound highly hydrophillic which precludes BBB penetration. Other dopamine prodrugs have been developed which seek to increase CNS penetration by increasing the amphipathic nature of the drug. For example, this carbamate ester dopamine prodrug has demonstrated exceptional BBB penetration.

In conclusion, N-coumaroyldopamine has great marketing potential for companies who are trying to eliminate DMAA from their products due to the recent en vitro studies. From a pharmacological standpoint, it will undubitably fall on its face in light of the hundreds of pharmaceutical papers which have examined similar compounds for the last 50 years.
References:
(1) http://www.pl.barc.usda.gov/downloads/jp41.pdf
(2) http://www.fasebj.org/content/19/6/497.full.pdf


